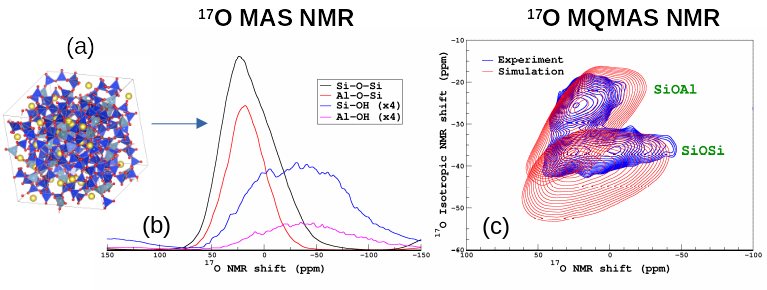
Because of the lack of long-range order, the structure of amorphous materials such as glasses remains difficult to study. Among the spectroscopic approaches that can be used, magic-angle sample spinning nuclear magnetic resonance (MAS NMR) has firmly established itself as one of the most powerful technique to determine the local environment of atoms allowing the building block of the glass network to be determined and quantified, such as SiO4, AlO4, BO4 and BO3 in aluminoborosilicate glasses (which is the base composition of nuclear waste glasses) with 29Si, 27Al and 11B NMR, respectively. With the help of isotopic enrichment, 17O MAS NMR can reveal essential information such the mixing between the network formers (Al, B, Si) and the organization and role of the network modifiers (Na,Ca). With the increase of computation resources, NMR can now be combined efficiently with Molecular Dynamics Simulations (MD) with the help of DFT calculations (the GIPAW method [1,2]). From MD structural models, NMR properties of each atom can be computed and NMR spectra can be simulated for a direct comparison with the experiments. On the one hand, this approach offers an effective way of assessing quality of MD simulations, on the other hand, NMR fingerprints of specific environments can be identified and this can greatly improve the interpretation of the NMR data. Several recent applications of the so-called MD-GIPAW to glass structure elucidation can be found in Refs. [3–6]
In the context of chemical durability of nuclear waste glasses, methodologies based on selective oxygen-17 enrichment of the alteration layer that form on the glass surface under aqueous or atmospheric alteration has been developed.[7] Such an approach is complementary to the use of proton NMR (1H) to selectively probe the alteration layer, which structural evolution is not completely understood. The latter can be seen as a hydrated nano-porous aluminosilicate materials that can retain residual amount of boron and sodium (those elements are generally used a tracer of the alteration kinetics as being leachable). However, in order to help the interpretation of the NMR spectra, the application of the MD-GIPAW methodology is still limited because of the difficulty to generate structural models of hydrated nano-porous gels by molecular Dynamics. Several recent approaches have been recently proposed. Generally, one builds initial materials with a controlled porosity,[8] insert water molecules and then relax the obtained structures. Because of the proton, sophisticated potentials such as reactive potentials (ReaxFF),[9, 10] or diffusive charge description of the proton (DCRP) are necessary.[11, 12]
In this work, we have generated several models of hydrous sodium and calcium aluminosilicate gels using the charge scaling procedure,[8, 13] where the Coulombic interactions are used to control the pore morphologies. Three potentials (two reactive ReaxFF, DCRP and a rigid ionic potential PMMCS, [14, 15]) were then used in a melt-quench approach to produce the final structural models of gels. The MD-GIPAW could then be applied to model the NMR spectra and compared with recent experiments on simplified nuclear glasses, using oxygen-17 NMR experiments (among others). Interpretation of 1H is also greatly improved with the help of such models, as will be discussed.
Nevertheless, MD-GIPAW calculations are severely limited in system size by the high-computational cost of the DFT computations. Typically systems up to 800 atoms can be considered with current HPC resources. A similar dilemma arises the choice of force-fields in MD simulations. Classical MD based on empirical models of interatomic interactions enable fast and scalable computations but at the expense of the accuracy permitted by ab initio (DFT) methods that are however computationally expensive. To solve this dilemma of accuracy versus efficiency, machine learning (ML) approaches have recently emerged as a powerful method for accelerating MD simulations and computing materials properties with an accuracy close to that of DFT methods.
This approach was pioneered by Behler and Parrinello [16, 17] (for recent reviews, see for example Refs [18, 19]) which made use of a deep neural network (NN) to represent the potential energy. The key of their approach was the introduction of so-called atomic descriptors that map the Cartesian coordinates of the atoms in the surroundings of a central atom into symmetry adapted functions used as inputs of the NN. As an alternative, Kernel Ridge Regression (or Gaussian Processes) can be used and lead to the Gaussian Approximation Potential (GAP) approach that is also very popular and found several applications to glasses. Those methods will be illustrated with application to the prediction of NMR properties [20] with the appealing perspectives of scaling up the MD-GIPAW methodology to models of several thousands of atoms or to include at affordable cost the impact of the finite temperature (mobility) in computed NMR spectra.
1. Pickard CJ, Mauri F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys Rev B. 2001;63(24):245101. https://doi.org/10.1103/PhysRevB.63.245101
2. Charpentier T, Menziani MC, Pedone A. Computational simulations of solid state NMR spectra: a new era in structure determination of oxide glasses. RSC Adv. 2013;3(27):10550–10578. https://doi.org/10.1039/C3RA40627J
3. Ishii Y, Salanne M, Charpentier T, Shiraki K, Kasahara K, Ohtori N. A DFT-Based Aspherical Ion Model for Sodium Aluminosilicate Glasses and Melts. J Phys Chem C. 2016;120(42):24370–24381. https://doi.org/10.1021/acs.jpcc.6b08052
4. Bisbrouck N, Bertani M, Angeli F, et al. Impact of magnesium on the structure of aluminoborosilicate glasses: A solid-state NMR and Raman spectroscopy study. J Am Ceram Soc. 2021;104(9):4518–4536. https://doi.org/10.1111/jace.17876
5. Gambuzzi E, Pedone A, Menziani MC, Angeli F, Florian P, Charpentier T. Calcium environment in silicate and aluminosilicate glasses probed by 43Ca MQMAS NMR experiments and MD-GIPAW calculations. Solid State Nucl Magn Reson. 2015;68–69:31–36. https://doi.org/10.1016/j.ssnmr.2015.04.003
6. Gambuzzi E, Pedone A, Menziani MC, Angeli F, Caurant D, Charpentier T. Probing silicon and aluminium chemical environments in silicate and aluminosilicate glasses by solid state NMR spectroscopy and accurate first-principles calculations. Geochim Cosmochim Acta. 2014;125:170–185. https://doi.org/10.1016/j.gca.2013.10.025
7. Narayanasamy S, Jollivet P, Jégou C, et al. Borosilicate glass alteration in vapor phase and aqueous medium. Npj Mater Degrad. 2022;6(1):1–15. https://doi.org/10.1038/s41529-022-00298-2
8. Beckers JVL, de Leeuw SW. Molecular dynamics simulation of nanoporous silica. J Non-Cryst Solids. 2000;261(1):87–100. https://doi.org/10.1016/S0022-3093(99)00607-9
9. Mahadevan TS, Du J. Atomic and micro-structure features of nanoporous aluminosilicate glasses from reactive molecular dynamics simulations. J Am Ceram Soc. 2021;104(1):229–242. https://doi.org/10.1111/jace.17465
10. Mahadevan TS, Sun W, Du J. Development of Water Reactive Potentials for Sodium Silicate Glasses. J Phys Chem B. 2019;123(20):4452–4461. https://doi.org/10.1021/acs.jpcb.9b02216
11. Mahadevan TS, Garofalini SH. Dissociative Water Potential for Molecular Dynamics Simulations. J Phys Chem B. 2007;111(30):8919–8927. https://doi.org/10.1021/jp072530o
12. Mahadevan T, Baroni A, Taron M, Gin S, Du J, Delaye J-M. Development of potentials for molecular dynamics simulations of dry and hydrated calcium aluminosilicate glasses by force matching and refinement. J Non-Cryst Solids. 2022;592:121746. https://doi.org/10.1016/j.jnoncrysol.2022.121746
13. Rimsza JM, Du J. Structural and Mechanical Properties of Nanoporous Silica. J Am Ceram Soc. 2014;97(3):772–781. https://doi.org/10.1111/jace.12707
14. Pedone A. Properties Calculations of Silica-Based Glasses by Atomistic Simulations Techniques: A Review. J Phys Chem C. 2009;113(49):20773–20784. https://doi.org/10.1021/jp9071263
15. Bertani M, Menziani MC, Pedone A. Improved empirical force field for multicomponent oxide glasses and crystals. Phys Rev Mater. 2021;5(4):045602. https://doi.org/10.1103/PhysRevMaterials.5.045602
16. Behler J. Perspective: Machine learning potentials for atomistic simulations. J Chem Phys. 2016;145(17):170901. https://doi.org/10.1063/1.4966192
17. Behler J, Parrinello M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Phys Rev Lett. 2007;98(14):146401. https://doi.org/10.1103/PhysRevLett.98.146401
18. Pedone A, Bertani M, Brugnoli L, Pallini A. Interatomic potentials for oxide glasses: Past, present, and future. J Non-Cryst Solids X. 2022;15:100115. https://doi.org/10.1016/j.nocx.2022.100115
19. Liu H, Fu Z, Yang K, Xu X, Bauchy M. Machine learning for glass science and engineering: A review. J Non-Cryst Solids X. 2019;4:100036. https://doi.org/10.1016/j.nocx.2019.100036
20. Chaker Z, Salanne M, Delaye J-M, Charpentier T. NMR shifts in aluminosilicate glasses via machine learning. Phys Chem Chem Phys. 2019. https://doi.org/10.1039/C9CP02803J